

Methods and Principles from our Scientific Staff
Immunoprecipitation Protocol
Required Materials
- Cell lysate
- Immobilized Protein A or Protein G (agarose or sepharose)
- Immunoprecipitation antibody
- Ice-cold lysis buffer
- Ice-cold PBS
- 2x SDS-PAGE sample loading buffer
- Centrifuge
- Rocking platform or rotator
- SDS-PAGE and Immunoblotting equipment/reagent
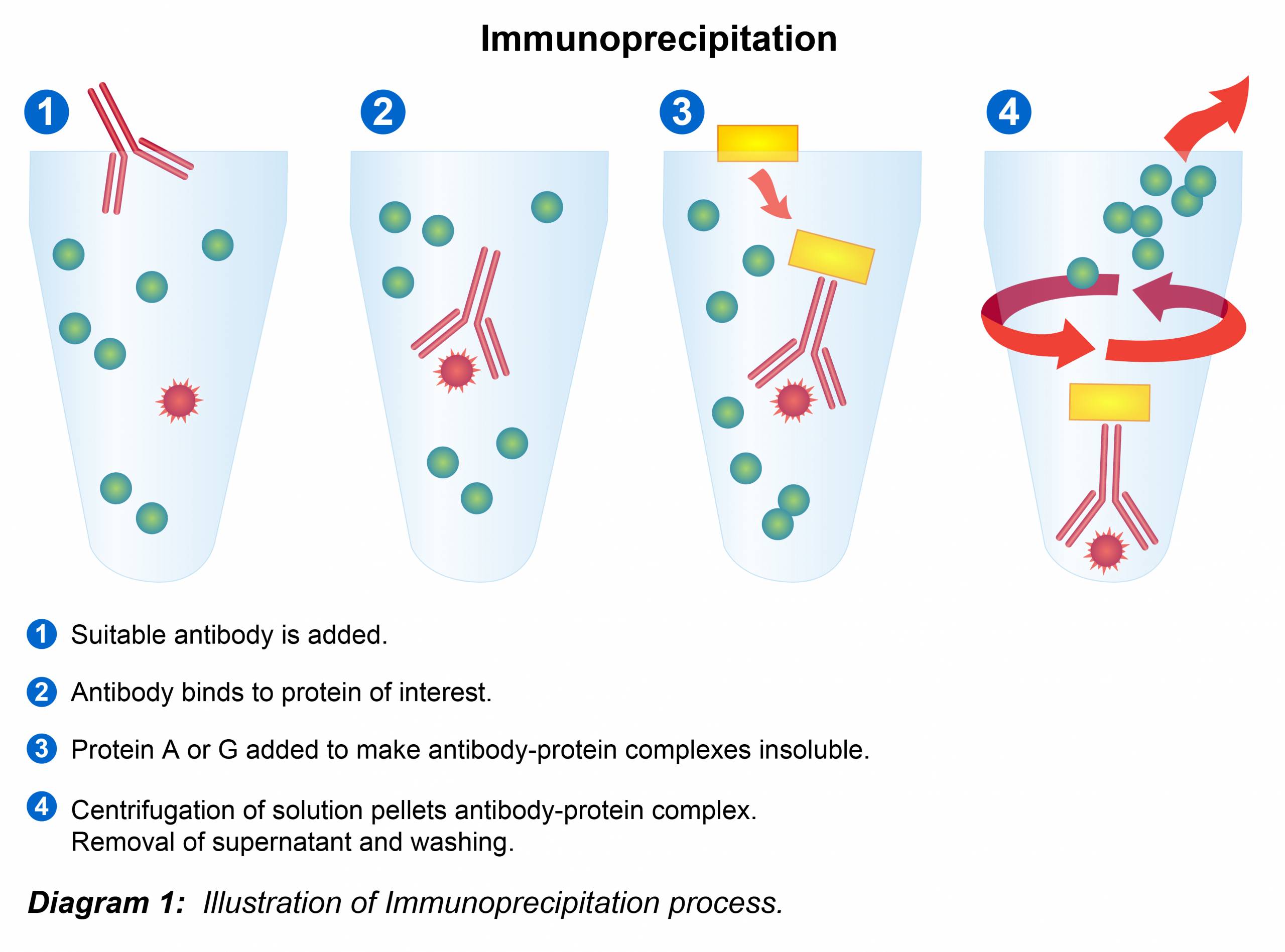
1.) Preparation of Lysates
Lysates from Cell Culture
Note: If using pre-existing cell lysate, proceed directly to Pre-clearing step.
Non-denaturing:
1. Place cell culture dish or flask on ice. Wash adherent cells twice with ice-cold PBS and drain off PBS. Wash non-adherent cells in PBS and centrifuge at 800 to 1000 rpm in a table-top centrifuge for 5 minutes to pellet the cells. Drain off PBS.
2. Add ice-cold lysis buffer (1ml per 107 cells/100mm dish/150cm2 flask; 0.5ml per 5×106 cells/60mm dish/75cm2 flask).
3. Scrape adherent cells off the dish using a cold plastic cell scraper then gently transfer the cell suspension into a pre-cooled microfuge tube.
4. Gently agitate the suspension on either a rocker or an orbital shaker at 4º C or on ice for 30 minutes to lyse cells.
5. Centrifuge the lysate at 14,000 x g in a precooled centrifuge for 15 – 30 minutes. Immediately transfer the supernatant to a fresh centrifuge tube on ice and discard the pellet.
Denaturing:
1. Add 100 ml denaturing lysis buffer per 0.5 to 2 x 107 cells.
2. Mix well by vortexing 2 to 3 seconds at maximum speed. Transfer the cell suspension to a microcentrifuge tube.
3. Heat samples to 95º C for 5 minutes to denature.
4. Dilute the suspension with 0.9 ml non-denaturing lysis buffer and mix gently.
(The excess 1% Triton X-100 in the nondenaturing lysis buffer quenches the SDS in the original denaturing buffer).
5. Fragment the DNA by passing the lysed suspension 5 to 10 times through a needle attached to a 1-ml syringe.
6. Incubate on ice or 4º for 5 minutes.
Lysates from Tissue
Note: Volumes of lysis buffer must be determined in relation to the amount of tissue present. Protein extract should not be too dilute to avoid loss of protein and to minimize the volume of samples to be loaded onto gels. The minimum concentration is 0.1 mg/ml; optimal concentration is 1–5 mg/ml. If denatured samples are required, use denaturing lysis buffer and perform steps 2 to 5 from the denaturing protocol above.
1. On ice, dissect the tissue of interest with pre-cooled tools. Work as quickly as possible to prevent degradation by proteases.
2. Place the tissue in round bottom microfuge tubes and immerse in liquid nitrogen to “snap freeze” and return tubes to ice.
3. For a ~5 mg piece of tissue, add ~300 µl lysis buffer to the tube, emulsify with an electric homogenizer.
4. Incubate for 2 hours at 4º C on an orbital shaker.
5. Centrifuge for 20 min at 12,000 rpm at 4º C in a microcentrifuge. Gently remove the tubes from the centrifuge and place on ice. Aspirate the supernatant and place in a fresh tube kept on ice, discard the pellet.
2.) Pre-clearing the Lysates
Note: The preclearing step is incorporated into the procedure to reduce the amount of non-specific contaminants in the cell lysate and to remove proteins with high affinity for Protein G or Protein A prior to the specific immunoprecipitation. The end result will be a lowering of background and an improved signal-to-noise ratio. However, if the final detection of the protein is by immunoblotting, pre-clearing may not be necessary, unless a contaminating protein is interfering with visualization of the protein of interest. Similarly, this step may be skipped in cases where the protein of interest is abundant in the sample.
1. To prepare Protein A or G agarose/sepharose, wash the beads twice with PBS and restore to a 50% slurry with PBS. It is recommended to cut the tip off of the pipette when manipulating agarose beads to avoid disruption them.
2. Pre-clear the cell lysate by adding 100 µl of either Protein A or G agarose/sepharose bead slurry per 1 mL of cell lysate and incubating at 4º C for 10-30 minutes on a rocker or orbital shaker.
3. Remove the Protein A or G beads by centrifugation at 14,000 x g at 4º C for 10 minutes. Transfer the supernatant to a fresh centrifuge tube and discard pellet.
3.) Immunoprecipitation
Note: Determine the protein concentration of the cell lysate (e.g. if performing a Bradford assay, dilute the cell lysate at least 1:10 before determining the protein concentration because of the interference of the detergents in the lysis buffer with the Coomassie-based reagent).
1. Dilute the cell lysate to approximately 1–10 µg/µl total cell protein with PBS. On ice, add 10–500 µg cell lysate plus the recommended amount of antibody (see antibody datasheet for recommended antibody concentration).
2. Incubate at 4º C for 1–2 hours or overnight on a rotator.
3. Capture the immunocomplex by adding 100 µl Protein A or G agarose/sepharose bead slurry (50 µl packed beads) and gently rocking on either a rocker or orbital shaker for either 1 hour or overnight at 4º C.
4. Centrifuge the tubes tube at 2,500 x g for 30 seconds at 4º C. Carefully remove the supernatant completely and wash the beads three to five times with 500 µl of ice-cold lysis buffer. Continue to pellet the beads in centrifuge between each wash. In order to minimize background, care should be given to remove the supernatant completely each time.
5. Resuspend the agarose/sepharose beads in 50 µl 2x sample loading buffer and mix gently.
6. Boil at 90–100º C for 5–10 minutes to dissociate the immunocomplexes from the beads.
7. Centrifuge the sample at 10,000 x g to pellet the beads.
8. Collect the supernatant carefully and load onto an SDS-PAGE gel to separate proteins prior to Western blot analysis. Alternatively, the supernatant samples can be collected, transferred to clean tube and frozen at -80º C for later use. Frozen supernatant should be reboiled for 5 minutes directly prior to loading on a gel.
Buffer Recipes
Non-denaturing lysis buffer
Use for antigens that are detergent soluble and can be recognized in native form by the antibody. Triton X-100 can be substituted for NP-40.
- 20 mM Tris HCl pH 8
- 137 mM NaCl
- 10% Glycerol
- 1% Nonidet P-40 (NP-40)
- 2 mM EDTA
Store up to 6 months at 4º C
Optional: Immediately before use add protease inhibitors.
RIPA (RadioImmunoPrecipitation Assay) buffer
More denaturing than NP-40 or Triton X-100 lysis buffer, RIPA buffer contains the ionic detergents SDS and sodium deoxycholate as active constituents and is particularly useful for nuclear membrane disruption for nuclear extracts. RIPA buffer gives low background but can denature kinases.
- 50 mM Tris HCl pH 8
- 150 mM NaCl
- 1% NP-40
- 0.5% sodium deoxycholate
- 0.1% SDS
Note: The 10% sodium deoxycholate stock solution (5 g into 50 ml) must be protected from light.
Detergent-free soluble protein lysis buffer
Some soluble proteins may not require use of detergents. Use this buffer with mechanical breakage of cells, e.g. repeated passage through a syringe or homogenization with a Dounce homogenizer. PBS containing:
- 5 mM EDTA >
- 0.02 % Sodium Azide
Store up to 6 months at 4º C
Optional: Immediately before use add protease inhibitors
Denaturing lysis buffer/buffer for non-detergent soluble antigens
Epitopes of native proteins are not accessible to antibodies that only recognise denatured proteins. When harvesting and lysing the cells, heat the cells in denaturing lysis buffer. This method can also be used for antigens that cannot be extracted from the cell with non-ionic detergents. Use of DNase1 will aid extraction of proteins from chromatin.
- 1% SDS
- 5 mM EDTA
Store up to 1 week at room temperature
Immediately before use add:
- 1 0mM dithiothreitol or beta-mercaptoethanol
- Protease inhibitors
- 15 U/ml DNase1
IP Wash Buffer
Washing the complexes can be done with RIPA, PBS or IP wash buffer. RIPA buffer is more stringent whereas PBS is less stringent.
- 10 mM Tris, Ph 7.4
- 1 mM EDTA
- 1 mM EGTA, pH 8.0
- 150 mM NaCl
- 1% Triton X-100
- 0.2 mM sodium ortho-vanadate
Optional: Protease inhibitor cocktail Protease Inhibitor Cocktail (100X):
- PMSF, 5 mg (50 µg/ml)
- Aprotinin, 100 µg (1µg/ml)
- Leupeptin, 100 µg (1µg/ml)
- Pepstatin, 100 µg (1µg/ml)
Phosphatase Inhibitor Cocktail (100X):
- 1 mM Na3VO4
- 1 mM NaF
Application Tips
1. The choice of lysis buffer depends upon the location of the protein (membrane, cytosolic or nuclear). Additionally, varying pH as well as detergent, salt and divalent cation concentrations are steps that can be taken to optimize specificity of the immunoprecipitation.
2. As soon as lysis occurs, proteolysis, dephosphorylation and denaturation begin. These events can be slowed down tremendously if samples are kept on ice or at 4º C at all times and appropriate inhibitors are added fresh to the lysis buffer. Mixtures (“cocktails”) of protease and phosphatase inhibitors are commercially available. If not using a cocktail, two of the most commonly used protease inhibitors for IP are PMSF (50 ug/ml) and aprotinin (1 ug/ml).
3. Immunoprecipitates allowed to incubate overnight may have a higher background compared to ones processsed for shorter periods of time due to time-dependent aggregation or denaturation of cellular proteins.
References
1. Bonifacino, J. S. et al. (2001) Immunoprecipitation. Curr. Protoc. Mol. Biol. Chapter 10:Unit 10.16.
2. Harlow, E. et al. (1999) Using Antibodies. Cold Spring Harbor Laboratory Press, NY.
